





What to Do If Cytotoxicity Test Results Are Positive?
The purpose of biocompatibility testing is to protect users from potential biological risks posed by medical devices. Every medical device, regardless of its classification, must evaluate cytotoxicity as long as it comes into contact with the human body. Due to the high sensitivity of cytotoxicity testing, combined with its low cost and short turnaround time, this evaluation is typically conducted through laboratory testing. However, because in vitro cytotoxicity testing is a sensitive detection method, positive results may sometimes occur.
ISO 10993-5:2009, "Biological evaluation of medical devices – Part 5: Tests for in vitro cytotoxicity," requires that all cytotoxicity results be carefully considered. It’s important to note that cytotoxicity testing only assesses the potential for cytotoxic effects in vivo through in vitro experiments and is not the sole method of evaluation. Medical devices are not used in vitro, so a positive in vitro cytotoxicity result alone cannot determine whether the device is unsuitable for a specific clinical application.
Therefore, any positive in vitro cytotoxicity result should be closely examined to determine whether there is a risk of in vivo cytotoxicity. A positive result does not automatically imply that the medical device lacks biocompatibility. After thorough research and biological risk assessment, it may still be possible to demonstrate the device's biological safety.
When positive results occur, it is generally recommended to follow these steps for evaluation:
Step 1: Confirm the Validity of the Test Results
Before investigating the cytotoxicity results, it is crucial to confirm the validity of the test results with the testing laboratory. This includes verifying whether the laboratory has ISO 17025 or GLP accreditation for in vitro cytotoxicity testing. These accreditations indicate that the laboratory has good quality control and that the staff is properly trained.
Additionally, confirm the following experimental details:
- Whether the correct extraction conditions and parameters were used;
- Whether all positive and negative controls were compliant and showed the expected viability;
- Whether expired products were used;
- Whether the test results showed good reproducibility;
- Whether the testing procedure was complete and no special sample preparation steps were omitted.
If the test was conducted in an ISO 17025 or GLP-accredited laboratory, it is generally assumed that these conditions have been met.
Step 2: Assess the Testing Parameters
If Step 1 confirms that the testing was conducted properly, the next step is to reassess the testing parameters used for in vitro cytotoxicity testing. For example, if previous tests on the same product showed no cytotoxicity, the manufacturer should assess whether the new test parameters and results comply with ISO requirements and whether the parameters and testing methods remained consistent between tests.
Furthermore, it is necessary to confirm whether the extraction conditions used for the medical device were reasonable. Typical extraction conditions are as follows:
- For devices in long-term or permanent contact with patients: extract at 37°C for 72 hours;
- For devices in short-term contact with patients: extract at 37°C for 24 hours;
- For devices in contact with patient skin or mucous membranes for less than 4 hours: extract at 37°C for 4 hours.
While the FDA generally does not recommend a 4-hour extraction or the use of a single solvent extraction, if there is a valid reason, the clinical application scenario can be considered when evaluating cytotoxicity.
Step 3: Evaluate the Materials and Components of the Medical Device
After confirming the validity of the test results and parameters, the next step is to evaluate the materials and components used in the device’s manufacturing process to determine whether any ingredients may have adverse effects that could lead to cytotoxicity. Certain metals, plasticizers, or cytotoxic inhibitors are known to cause cytotoxicity reactions.
If cytotoxicity is indeed observed, two follow-up approaches can be considered:
- If technically feasible, repeat the cytotoxicity test using a product that does not contain suspected cytotoxic materials/chemicals. If the result is negative (i.e., passed), the source of the cytotoxicity can be identified.
- Repeat the cytotoxicity test with a dilution solution and compare it with similar devices already on the market. If it can be demonstrated that the test device performs similarly to a device that has been safely marketed, then it is likely that no cytotoxicity will be observed under clinical use conditions.
This approach is sometimes used for products like liquids, gels, or textiles, which are typically difficult to test and often result in cytotoxic reactions.
Step 4: Evaluate Changes in the Process and Testing
If the potential cytotoxic material cannot be easily identified, compare the current data with historical data to identify similarities or differences. For example, were there any cytotoxic reactions in past tests, and what were the results?
If no cytotoxic reactions were previously observed, has there been a change in the manufacturing process (e.g., changes in suppliers, processes, or packaging)? Even small, seemingly insignificant changes in the process can affect the results of cytotoxicity testing. For example, if workers started handling products with latex gloves (latex is a common positive control in testing), previously non-cytotoxic devices may now show cytotoxic activity.
Step 5: Investigate Further with New Testing
If the source of the positive result cannot be easily identified, further investigation may be needed. For example, testing different batches of products can help determine whether the issue affects a specific batch or all batches. Alternatively, testing at different stages of production can help narrow down and ultimately identify the source of the cytotoxicity reaction.
Step 6: Address Cytotoxicity Failure in Biological Risk Assessment: Risk Assessment
If it is confirmed that the product does exhibit cytotoxicity and the source of the cytotoxicity is identified, it is recommended to perform a biological risk assessment. The risk assessment should summarize the tests that have been conducted and explain why the device’s cytotoxicity potential is suitable for the evaluation of its biocompatibility.
During the risk assessment, additional testing data may be required, such as determining the release kinetics of cytotoxic compounds to evaluate the potential toxicity to the patient during the device’s expected use.
Moreover, the risk assessment may integrate literature review, post-market surveillance data, additional chemical testing, and/or available in vivo data to demonstrate that no adverse reactions will occur during clinical use.
Email:hello@jjrlab.com
Write your message here and send it to us
 Toothbrush FDA Certification Testing
Toothbrush FDA Certification Testing
 Snoring Device FDA 510k Standard Testing
Snoring Device FDA 510k Standard Testing
 Single Use Intravenous Catheter Certification Test
Single Use Intravenous Catheter Certification Test
 Silicone Material Product Compliance Certification
Silicone Material Product Compliance Certification
 What to Do If Cytotoxicity Test Results Are Positi
What to Do If Cytotoxicity Test Results Are Positi
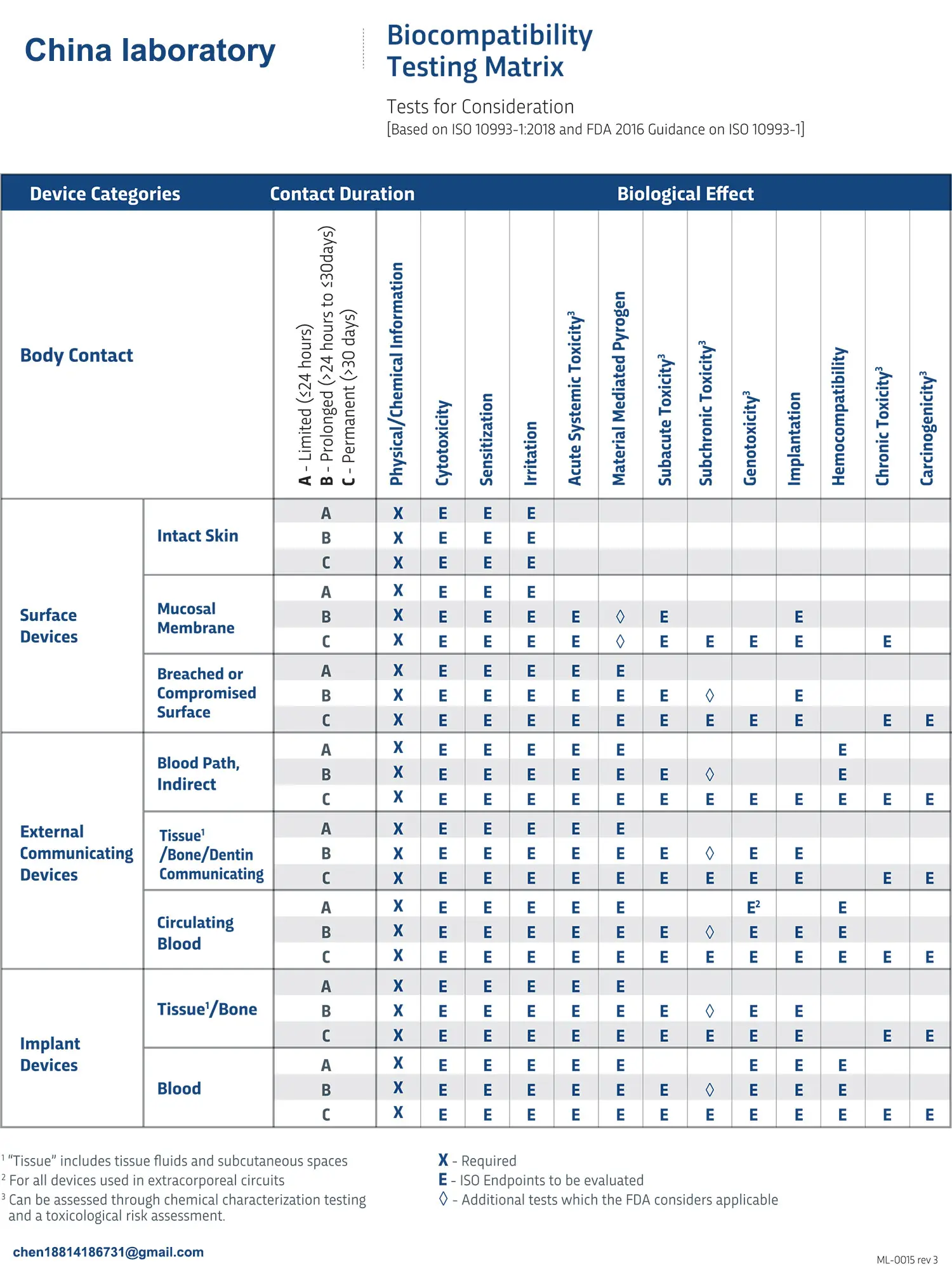 ISO 10993:5 Cytotoxicity Testing Methods
ISO 10993:5 Cytotoxicity Testing Methods
 FDA ISO 10993-1 Biocompatibility Evaluation Guidel
FDA ISO 10993-1 Biocompatibility Evaluation Guidel
 In Vitro Cytotoxicity Testing for Medical Devices
In Vitro Cytotoxicity Testing for Medical Devices
Leave us a message
24-hour online customer service at any time to respond, so that you worry!