





What Is CE Certification for Medical Devices?
Introduction to European CE certification
According to statistics from MedTec Europe, the European medical device market was approximately €135 billion in 2022, accounting for about 27% of the global market and ranking as the second-largest medical device market after the United States.

Since May 2021, medical device manufacturers must comply with the European Medical Device Regulation 2017/745 instead of the Medical Device Directive 93/42/EEC to obtain CE marking approval. Therefore, medical devices must be classified according to the MDR.
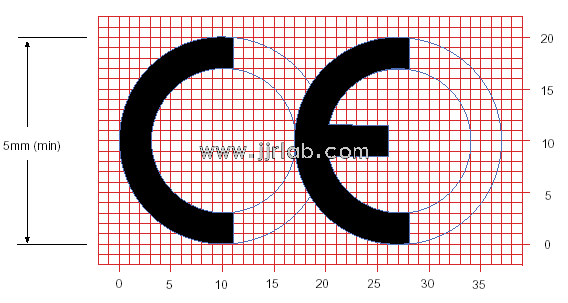
To demonstrate that your device complies with the essential requirements of these CE directives, you need to affix the CE mark to it. Your product needs to undergo the CE certification marking process. The direction of the latter depends on the category of your medical device and the qualified assessment route you choose. The specific characteristics of your medical device will determine its category and the level of risk to patients. For example, features such as intended use, invasiveness, and local or systemic effects.
Procedure and Process for Medical Device CE Certification
(I) Confirm Exporting Country
If exporting to the European Economic Area (EEA), including EU and European Free Trade Association (EFTA) member states, CE certification may be required.
(II) Confirm Product Category and Relevant EU Product Directives
According to the European framework, medical devices are divided into four classes: Class I, Class IIa, Class IIb, and Class III. Class III medical devices pose the highest risk. Today, due to stricter rules of the new regulatory system, the classification of many devices has changed. Previously classified as Class IIa or IIb, they are now categorized as Class III. If your medical device belongs to any category other than Class I, you must provide evidence to the certification body that your product meets the essential requirements of the corresponding CE directive.
(III) Designate an "EU Authorized Representative"
For manufacturers located outside the 30 EEA member countries, it is mandatory to designate an EU Authorized Representative to ensure the consistency of product safety throughout the distribution and use process in the European market. Technical files must be stored within the EU for inspection by supervisory authorities at any time. Remedial measures must be taken for products found to be non-compliant with CE requirements by market surveillance authorities or for products that have been labeled with CE marks but have experienced accidents during use.
(IV) Confirm Certification Required Mode (Module)
For almost all EU product directives, manufacturers are typically provided with several CE certification modules to choose from according to their specific circumstances. CE certification modules can generally be divided into nine basic modules.

(V) Adopting the "Self-Declaration" Mode or "Must Be Certified by a Third-Party Certification Body"
For products with a lower risk level, manufacturers may choose the "Internal Production Control (Self-Declaration)" mode for CE certification. Products with higher risk levels must involve a notified body (NB). Depending on the risk level, manufacturers must select other modes besides Mode A, or combine Mode A with other modes for CE certification.
(VI) Establish Technical Files and Their Maintenance and Updates
EU law requires that technical files for products labeled with CE marks and placed on the European market must be stored within the EU for inspection by supervisory authorities at any time. Technical files should be promptly updated if there are any changes. Technical documentation is a crucial requirement in EU medical device directives, aimed at ensuring that companies have sufficient technical information and evidence for use in inspections or litigation disputes. The requirements for "technical files" vary among EU directives. For example, the Medical Device Directive 93/42/EEC may include items such as quality manuals, company profiles, CE conformity declarations, product descriptions, risk analysis reports, and packaging and labeling descriptions.
Email:hello@jjrlab.com
Write your message here and send it to us
 ASTM D4169 Drop Test
ASTM D4169 Drop Test
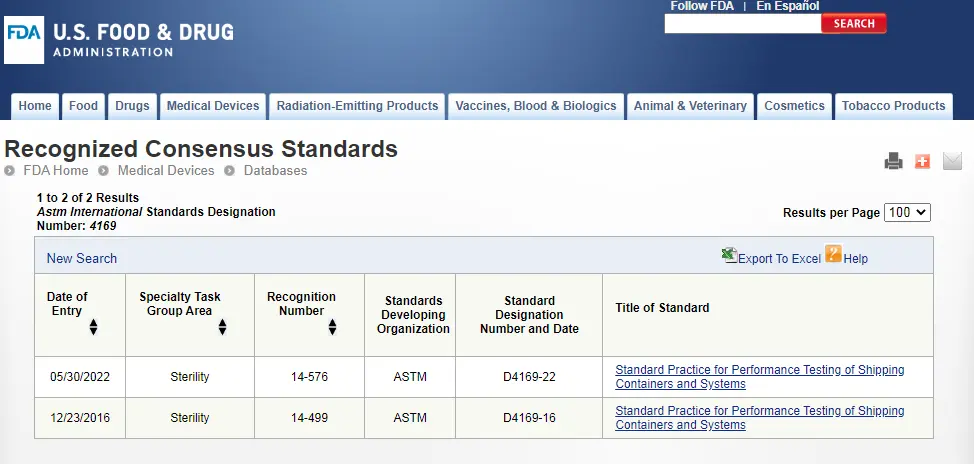 ASTM D4169 Packaging Simulation Transportation Tes
ASTM D4169 Packaging Simulation Transportation Tes
 What is ASTM D4169 Testing?
What is ASTM D4169 Testing?
 ASTM D4169-23 Test Standard Revision
ASTM D4169-23 Test Standard Revision
 Transport Simulation Testing for Medical Device Pa
Transport Simulation Testing for Medical Device Pa
 EU CE Certification Guidelines for Lighting Fixtur
EU CE Certification Guidelines for Lighting Fixtur
 Lithium Battery Export: CB Certification & IEC
Lithium Battery Export: CB Certification & IEC
 How to Apply for One FCC Certificate for Multiple
How to Apply for One FCC Certificate for Multiple
Leave us a message
24-hour online customer service at any time to respond, so that you worry!