





Medical Device CE Certification Cost
The cost of CE certification for medical devices typically ranges from approximately $8,000 to $12,000 USD for standard products. For more complex products, the cost can reach tens of thousands of dollars.
")
To sell medical devices in the EU market, manufacturers must comply with stringent regulatory requirements. CE certification is a crucial marker that ensures compliance with EU standards for health, safety, and environmental protection. Below is a detailed breakdown of the CE certification process:
Understand Applicable EU Regulations
• EU Medical Device Regulation (MDR) 2017/745: As of May 2021, all medical devices must comply with MDR, replacing the earlier Medical Device Directive (93/42/EEC) and In Vitro Diagnostic Directive (98/79/EC).
• In Vitro Diagnostic Regulation (IVDR) 2017/746: Applies to in vitro diagnostic products, emphasizing clinical evidence and risk assessment.
Determine the Product Classification
Medical devices in the EU are classified by risk category, which determines certification requirements. The main categories are:
• Class I (low risk): Self-certification is allowed without a Notified Body's approval.
• Class IIa and IIb (medium risk): Require review by a Notified Body.
• Class III (high risk): Must undergo Notified Body review with stringent clinical data and evaluation.
Classification depends on the intended use, degree of contact with the human body, and associated risk assessment.
Conformity Assessment
Conformity assessment ensures the product meets EU regulatory requirements. Based on the classification, manufacturers must complete the following:
• Technical Documentation: Prepare detailed technical files covering design, manufacturing, clinical evaluation, and risk management.
• Clinical Evaluation and Trials: For high-risk devices, clinical trial data must demonstrate safety and efficacy. Low-risk devices may rely on literature reviews or existing data.
• Risk Management: Evaluate potential risks and implement corresponding control measures.
Choose a Notified Body
Depending on the risk category, manufacturers may need a Notified Body to audit and certify the product:
• Class I: No Notified Body involvement required.
• Class II and III: Select an EU-accredited Notified Body to review technical documentation and quality management systems (e.g., ISO 13485).
The choice of Notified Body depends on the product category and complexity. Manufacturers should ensure the selected body is qualified and experienced in medical device certification.
Quality Management System (QMS)
• ISO 13485: The standard for medical device quality management systems. Most medium- to high-risk products must comply with ISO 13485. The Notified Body will audit the manufacturer’s production and management processes.
Before CE certification, manufacturers must establish and maintain an effective QMS to ensure quality control throughout product design and production.
Clinical Data and Evaluation
• Clinical evaluation is mandatory for all medical devices and includes:
§ Clinical Trials: To prove safety and efficacy for the intended use.
§ Existing Clinical Data: Low-risk devices may use existing data to support safety and effectiveness.
Class III products require clinical trials and a detailed clinical evaluation report.
Technical Documentation and Declaration of Conformity
• Technical Documentation: Includes detailed descriptions of product design, risk assessments, performance tests, and clinical data.
• Declaration of Conformity (DoC): Manufacturers issue a DoC stating the product meets all EU regulatory requirements and submit it during CE certification.
Use of the CE Mark
• Once certified, manufacturers may affix the CE mark to their product, signifying compliance with EU regulations.
• Any modifications or updates to the product must ensure continued compliance with the CE certification.
Market Surveillance and Post-Market Supervision
• EEA Market Surveillance: Manufacturers must ensure ongoing compliance after market entry. EU member states conduct periodic inspections to enforce regulatory adherence.
• Incident Reporting: Manufacturers must report adverse events (e.g., product defects or usage risks) to the European Medicines Agency (EMA) or national regulators and implement corrective actions.
Key Changes in MDR Compliance
The new MDR imposes stricter requirements compared to the previous directive, including:
• Enhanced monitoring throughout the product lifecycle.
• Emphasis on clinical data and trials, especially for high-risk devices.
• Increased post-market tracking requirements, such as "post-market surveillance" and "incident reporting."
• Unique Device Identification (UDI) for all medical devices to facilitate traceability.
CE certification is a vital step to ensure medical device compliance, encompassing all stages from design and clinical evaluation to market supervision. Manufacturers must understand and adhere to EU regulations (MDR/IVDR) to ensure safety, efficacy, and compliance. Achieving CE certification not only allows access to the EU market but also enhances international competitiveness.
Email:hello@jjrlab.com
Write your message here and send it to us
 Toothbrush FDA Certification Testing
Toothbrush FDA Certification Testing
 Snoring Device FDA 510k Standard Testing
Snoring Device FDA 510k Standard Testing
 Single Use Intravenous Catheter Certification Test
Single Use Intravenous Catheter Certification Test
 Silicone Material Product Compliance Certification
Silicone Material Product Compliance Certification
 What to Do If Cytotoxicity Test Results Are Positi
What to Do If Cytotoxicity Test Results Are Positi
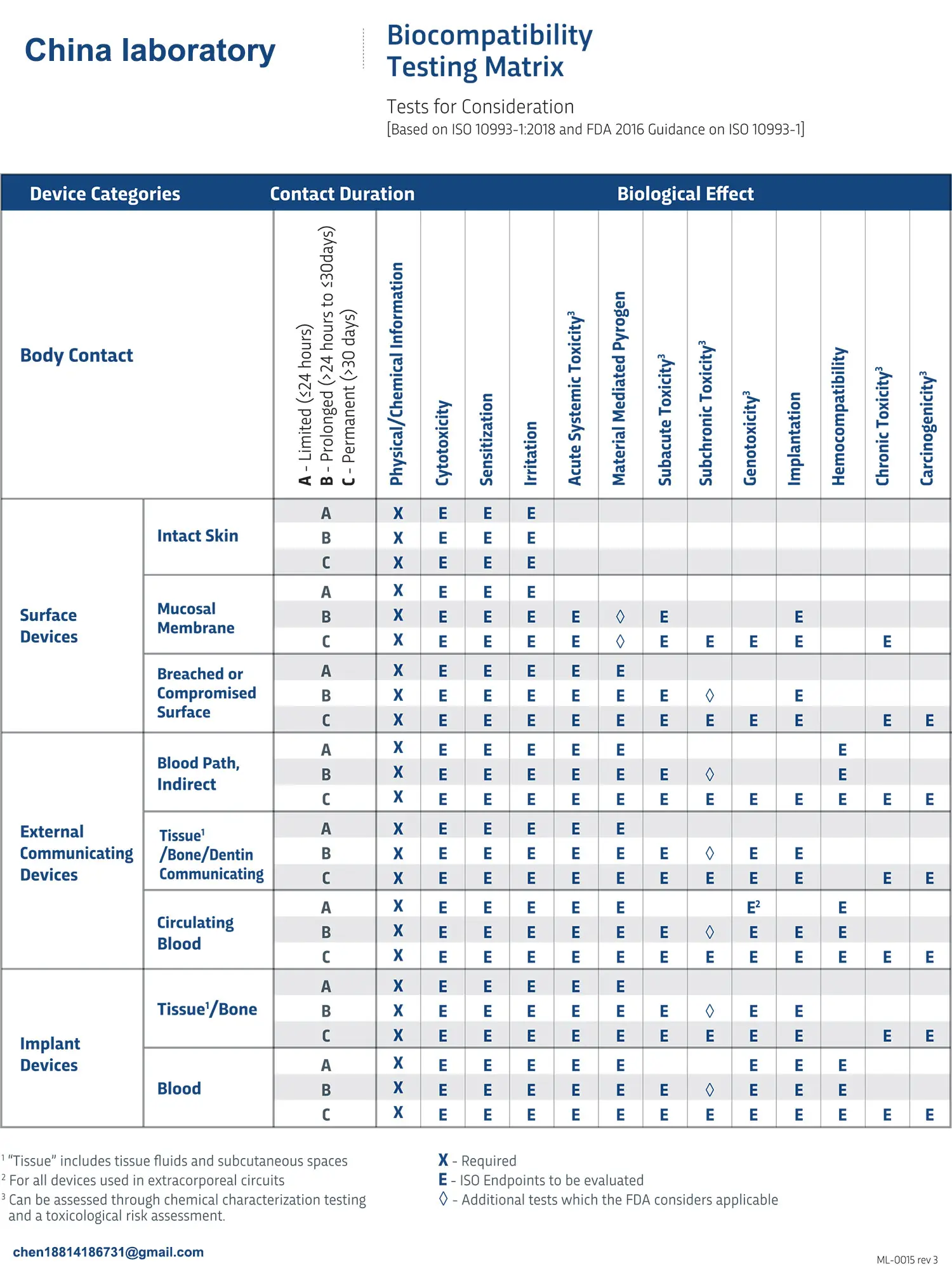 ISO 10993:5 Cytotoxicity Testing Methods
ISO 10993:5 Cytotoxicity Testing Methods
 FDA ISO 10993-1 Biocompatibility Evaluation Guidel
FDA ISO 10993-1 Biocompatibility Evaluation Guidel
 In Vitro Cytotoxicity Testing for Medical Devices
In Vitro Cytotoxicity Testing for Medical Devices
Leave us a message
24-hour online customer service at any time to respond, so that you worry!