





ISO 10993-1 Biological Evaluation and Chemical Characterization
What is the difference between ISO 10993-1 and submitting documents for FDA review?
In June 2016, the U.S. FDA officially announced the biological evaluation standard “Use of International Standard ISO 10993-1 , Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process”
The FDA's upgraded regulations differ from ISO 10993-1 in some test evaluation and extraction requirements, especially for high-risk degradable implant products, which require observation of the short-, medium-, and long-term effects of sample degradation after implantation on the organism, and for the quality control of pyrogen and endotoxin testing of implants or non-pyrogen products.
In addition, the FDA does not recognize the ISO 17025 system and requires that biocompatibility testing must be completed in a formal GLP biological laboratory. Therefore, medical device manufacturers who want to apply for US FDA registration must entrust a laboratory with GLP qualifications to complete relevant biological tests in accordance with FDA biological evaluation standards.
The original text of the FDA regulations is excerpted as follows:
Any in vitro or in vivo biological safety experiments or tests should be conducted in accordance with recognized Good Laboratory Practice (GLP) regulations including, but not limited to, the assignment of competent trained staff in the conduct of biocompatibility testing.(hello@jjrlab.com)
Is there a distinction between extractables and degradation products of a material?
Extractables refer to substances released from products (materials, components or devices) after solvent extraction; degradation products are particles or chemical substances produced by chemical decomposition of the original materials in the body, so the definitions of the two are different. The biggest difference after the revision of ISO 10993-1 is that it is necessary to consider comprehensive information of product materials before the test is carried out, and then conduct biological tests after risk assessment and chemical characterization evaluation (obtaining leachable information and toxicological assessment information).
Is it possible to perform sub-chronic systemic toxicity testing on dental materials without using non-polar extraction?
Currently, ISO 10993-1 regulations require that biological tests be conducted using both polar and non-polar solvents for leaching and testing, so the subchronic toxicity of dental materials also requires the evaluation of the toxicity of both extracts. The ISO 7405 dental series standard mentions that subchronic toxicity is recommended to be administered orally, so it is recommended to refer to this standard for dental product registration.
When a product is used repeatedly and each use is not destined for the same patient, how should the cumulative contact time and dosage be considered?
The revised version of ISO 10993-1 specifically mentions that the risk of reusable medical devices should be assessed based on the cumulative time the product is in contact with the patient. This requirement will be calculated based on the contact time of a single patient. If the same patient is not specified, the biological risk assessment can be conducted based on the actual clinical use situation.
Why do products that are provided non-sterile need to be sterilized before cytotoxicity testing?
Cytotoxicity tests often cause false positive results due to interference from microorganisms. Therefore, when preparing samples, it is important to ensure the cleanliness of the environment and the integrity of the product packaging to reduce the risk of failure in cytotoxicity tests. If the product is a non-sterile medical device after it is launched on the market, it is recommended to simulate the actual market status of the product and perform a simple cleaning before sending it for testing. If you are worried that the product has microorganisms that need to be sterilized, you need to evaluate the impact of sterilization on the material.
What is the specific process of toxicological evaluation? Can you give an example? Can it be carried out by using the biological risk evaluation in risk management?
After the release of ISO 10993-1:2018, chemical characterization analysis of materials is the first step in biological evaluation, and the allowable limit of leachable substances needs to be derived, toxicological evaluation and risk control need to be performed. The specific process is based on Figure 1 (derived from Figure 1 of ISO 10993-17). Toxicological evaluation is recommended to be conducted by professional toxicologists; risk management methods are recommended to refer to ISO 15499 and ISO 14971 standards.

If conditions do not allow the use of sterilized products for testing, can key materials be used instead?
According to the requirements of ISO 10993 biological standards, the final product or samples made of the same material as the final product (including sterilization method) must be used for testing; if only key material testing is used, a reasonable explanation must be given.
Drug-eluting stents, testing cytotoxicity, is it necessary to conduct drug-containing testing in case of acute toxicity? Does drug-containing testing have any impact on acute toxicity?
Drug-containing devices need to first confirm the safety of the drug and the drug compatibility with the material, and then conduct biological tests as final products in accordance with regulations. If you are concerned about the impact of the drug on the biological test, you can collect drug-related safety data and literature before conducting the test.
If the cytotoxicity test of a band-aid product shows potential cytotoxicity, what is the next step?
Band-aid products are easily affected by colloids in cytotoxicity tests. It is recommended to collect relevant literature or compare the test results with those of similar samples already on the market: if the cytotoxicity is mild (1 point), it meets the regulatory requirements (cell survival rate must be above 70%); if the toxicity score is high, it is recommended to replace it with other medical-grade materials that have been verified by biological tests.
What are the differences among acute, subacute, subchronic and chronic poisons? How should we choose according to the product?
The differences in the selection of these systemic toxicity tests are based on the evaluation of the site and time of product contact with patients. If the product contacts tissue mucosa or in vitro connected devices, and the contact time is less than 24 hours, only acute toxicity tests are required; for medical devices that are in contact for more than 30 days, in addition to acute toxicity, subchronic toxicity or chronic toxicity tests are required. Acute systemic toxicity only observes the short-term toxic reactions of the product, while subchronic and chronic toxicity mainly observe the animals after continuous feeding and sacrifice, and use pathological dissection to confirm whether there are lesions in tissues and organs. If there is no relevant literature to support the safety of the material (such as new materials), chronic toxicity tests need to be evaluated; conversely, if there is relevant safety data, it can be exempted.
Is it necessary to use both sexes when selecting experimental animals in biological experiments (the equipment itself does not distinguish between sexes when used)?
The selection of animals for biological tests must be based on the requirements of the ISO 10993 test standard. Some tests require single-sex testing (such as sensitization) and some tests require dual-sex testing (such as subacute/subchronic toxicity testing). The corresponding test items can be evaluated based on the intended use and contact time of the product.
If the supplier provides chemical characterization, can the device manufacturer be exempted from chemical characterization? If the supplier cannot provide it, does the device manufacturer have to do the testing itself?
If the supplier provides relevant chemical characterization information of the material, it can be cited in the product risk assessment report. However, the device manufacturer also needs to evaluate the risks of additives and residues in the product production process. If the supplier can provide relevant information, the risk of test failure can be reduced. If the supplier cannot provide relevant reports, the manufacturer needs to carry out the material chemical characterization risk assessment plan on its own.
For products that have been used for a long time for more than 30 days, “sub-chronic toxicity has been tested”. Is it necessary to test for acute systemic toxicity, or is it optional?
According to the 10993-1 evaluation form, products used for long periods of time must undergo acute systemic toxicity tests in addition to subchronic toxicity tests. Acute systemic toxicity is the observation of the short-term immediate toxic reaction of the product after it comes into contact with an organism, and it differs from the indicators used in subchronic toxicity assessments.
What is the difference between 15499 and 14971? Do I need to cite 15499 in my biological review?
ISO 14971 and ISO 15499 both describe risk management methods. 14971 talks about the basis for risk management in the life cycle of medical devices, while 15499 is a risk assessment process for biocompatibility. Both of them mention how to use risk management methods to conduct biocompatibility assessments. However, risks are everywhere, so manufacturers are advised to combine the two standards to conduct product risk assessments.
Choice of extraction conditions, choice of temperature, choice of specific surface area?
According to the usage and product type of the medical device, the extraction conditions such as temperature and surface area ratio can be selected according to the requirements of ISO 10993-12: Sample preparation and reference materials. The extraction method for material chemical characterization can also refer to this standard.
How should the biological evaluation be conducted when changing the manufacturer of solvent cyclohexanone?
If the solvent manufacturer is changed, a new risk assessment analysis is required (changing the solvent is a major change that will affect product quality). After the assessment, if biological testing is required to confirm product safety, the corresponding experiments will be carried out and the test results will be summarized in the biological risk assessment report.
Email:hello@jjrlab.com
Write your message here and send it to us
 ASTM D4169 Drop Test
ASTM D4169 Drop Test
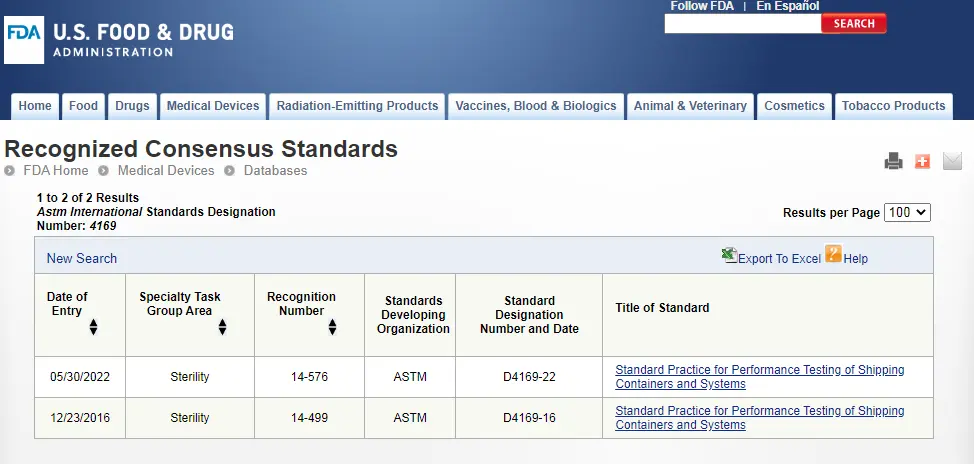 ASTM D4169 Packaging Simulation Transportation Tes
ASTM D4169 Packaging Simulation Transportation Tes
 What is ASTM D4169 Testing?
What is ASTM D4169 Testing?
 ASTM D4169-23 Test Standard Revision
ASTM D4169-23 Test Standard Revision
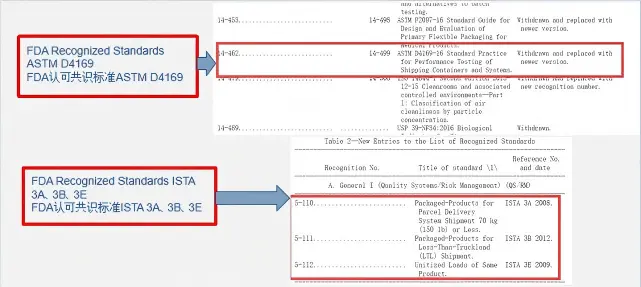 Transport Simulation Testing for Medical Device Pa
Transport Simulation Testing for Medical Device Pa
 EU CE Certification Guidelines for Lighting Fixtur
EU CE Certification Guidelines for Lighting Fixtur
 Lithium Battery Export: CB Certification & IEC
Lithium Battery Export: CB Certification & IEC
 How to Apply for One FCC Certificate for Multiple
How to Apply for One FCC Certificate for Multiple
Leave us a message
24-hour online customer service at any time to respond, so that you worry!