





FDA ISO 10993-1 Biocompatibility Evaluation Guidelines
This text provides a detailed comparison and analysis of the ISO 10993 series standards and the FDA biocompatibility evaluation guidelines. It clearly outlines key issues in the biocompatibility evaluation process, such as risk management, test sample preparation, and testing requirements. To make the structure clearer and more comprehensible, I will reorganize and format this content:
Overview of ISO 10993 Series Standards
The ISO 10993 series includes more than twenty individual standards that cover various biological evaluation metrics, sample preparation methods, and testing approaches for specific materials. Among them, ISO 10993-1 — Biological Evaluation of Medical Devices – Part 1: Evaluation and Testing within a Risk Management Process — outlines the fundamental principles of biocompatibility evaluation and assists in planning the evaluation framework, making it the core of the series.
")
With the significant updates in the 2018 edition of ISO 10993-1, the FDA issued a draft revision of its biocompatibility evaluation guidelines in October 2020, which was followed by the final guideline release in September 2023, replacing the 1995 G95-1 Blue Book Memo.
Similarities and Differences Between ISO 10993-1 and the FDA’s New Biocompatibility Evaluation Guidelines
The FDA’s revised biocompatibility evaluation guidelines largely adopt the principles of ISO 10993-1, following a risk management process for biological evaluations. However, the FDA made several adjustments and did not fully adopt the ISO 10993-1:2018 standard, particularly in terms of the necessity of evaluating the physical and chemical properties of medical devices, biocompatibility indicators, and key points for biocompatibility testing. This article focuses on analyzing the FDA’s approach and considerations regarding biocompatibility evaluation for medical devices within a risk management framework.
Risk Management-Based Biocompatibility Evaluation
1. Risk Assessment of Medical Devices
Risk assessment is a critical initial stage in the risk management process, including both risk analysis and risk evaluation. Risk analysis systematically uses existing information to identify potential hazards and estimate risks — i.e., the likelihood and severity of harm. Risk evaluation compares estimated risks with acceptable risk standards to determine whether the risk is acceptable and identifies the risks that require mitigation.
The FDA emphasizes that, for medical device risk assessment, the final product form should be the basis for evaluation, considering factors such as the materials used, manufacturing process, sterilization method, and any residues from processing aids. It is also important to account for the device's intended clinical use, such as the target site, exposure time, and expected user population.
2. Identification of Potential Biocompatibility Risks
The core of the biological evaluation is understanding the overall exposure of the human body to the medical device. For devices containing potentially toxic chemicals, safety evaluations should include an analysis of material components, expected additives, processing contaminants, residues, and leachable substances for potential toxic and chemical risks. In some cases, chemical analysis may be used to support decisions regarding whether biocompatibility testing is necessary, especially for toxicological risk assessment of chemicals.
However, chemical analysis alone is not sufficient to identify all biocompatibility risks. The FDA specifically mentions that the physical characteristics of the device (e.g., surface roughness, geometry, mechanical forces) should also be considered in the risk assessment process.
3. Identifying and Mitigating Risks Based on Available Information
To reduce unnecessary testing and adhere to the 3Rs principle (Replacement, Reduction, Refinement), the FDA encourages making full use of all available relevant information during biocompatibility evaluation. Key sources of information for evaluation include:
- Literature and public data: Reviewing existing toxicity literature and other publicly available information to confirm the toxicity risks of material components.
- Clinical experience: Using existing clinical data to assess whether additional biocompatibility testing is needed.
- Animal testing experience: In vivo animal testing can replace certain biocompatibility tests, particularly when relevant experience exists.
- Recognized standards: Established standards for specific medical devices or materials assist in risk evaluation.
- Previous review experience: FDA recognizes prior review experiences, such as data from previous or comparable products.
Considerations for Biocompatibility Testing
The updated ISO 10993-1:2018 standard no longer specifies mandatory biocompatibility tests based on device risk. Instead, it recommends using biological evaluation metrics, which may include reviewing available information or conducting a series of material, physical, and chemical evaluations to justify the lack of additional testing data.
1. cytotoxicity testing
The FDA recommends using solvents that can dissolve both polar and nonpolar components of the test material, with extraction times between 24 to 72 hours. In certain cases, if the device contains new materials, cytotoxicity testing may require a combination of direct contact and elution methods.
2. Sensitization Testing
The FDA accepts two methods for sensitization testing: the guinea pig maximization test and the mouse local lymph node assay (LLNA). Additionally, the FDA emphasizes using validated, standardized methods for LLNA testing and selecting the appropriate testing method based on the physical-chemical characteristics of the material.
3. Hemocompatibility Testing
For devices that come into direct contact with circulating blood, the FDA recommends hemolysis, complement activation, and thrombosis testing. For devices that only have indirect contact with blood, hemolysis testing is sufficient. For novel materials not yet used in approved devices, an in vitro thrombosis evaluation may be necessary.
4. Pyrogenicity Testing
For implantable devices or sterile devices that come into direct or indirect contact with the cardiovascular system, lymphatic system, cerebrospinal fluid, etc., the FDA recommends compliance with pyrogen limit specifications.
5. Implantation Testing
The FDA suggests that implantation tests, which simulate clinical conditions, are more suitable for evaluating high-risk implantable devices to understand their effects on local and systemic tissues.
6. Genotoxicity Testing
For devices that come into contact with human tissues for more than 24 hours, the FDA recommends performing genotoxicity tests. These should include bacterial gene mutation tests and in vitro mammalian genotoxicity assays.
7. Carcinogenicity Testing
The FDA recommends carcinogenicity evaluations for devices that have prolonged contact with human tissues. For new materials not yet used in approved devices, potential carcinogenicity may need to be analyzed through structure-activity relationship models.
Through these well-designed evaluation and testing methods, the FDA ensures that biocompatibility evaluations of medical devices more accurately reflect the potential risks to patient safety. At the same time, it promotes more rational alternative testing methods to reduce animal testing, advancing the safety and effectiveness of medical devices.
If your product requires ISO 10993 biological testing, JJR Laboratory in China can provide these services, with the standard three biocompatibility tests costing around $4,000 USD.
Email:hello@jjrlab.com
Write your message here and send it to us
 Toothbrush FDA Certification Testing
Toothbrush FDA Certification Testing
 Snoring Device FDA 510k Standard Testing
Snoring Device FDA 510k Standard Testing
 Single Use Intravenous Catheter Certification Test
Single Use Intravenous Catheter Certification Test
 Silicone Material Product Compliance Certification
Silicone Material Product Compliance Certification
 What to Do If Cytotoxicity Test Results Are Positi
What to Do If Cytotoxicity Test Results Are Positi
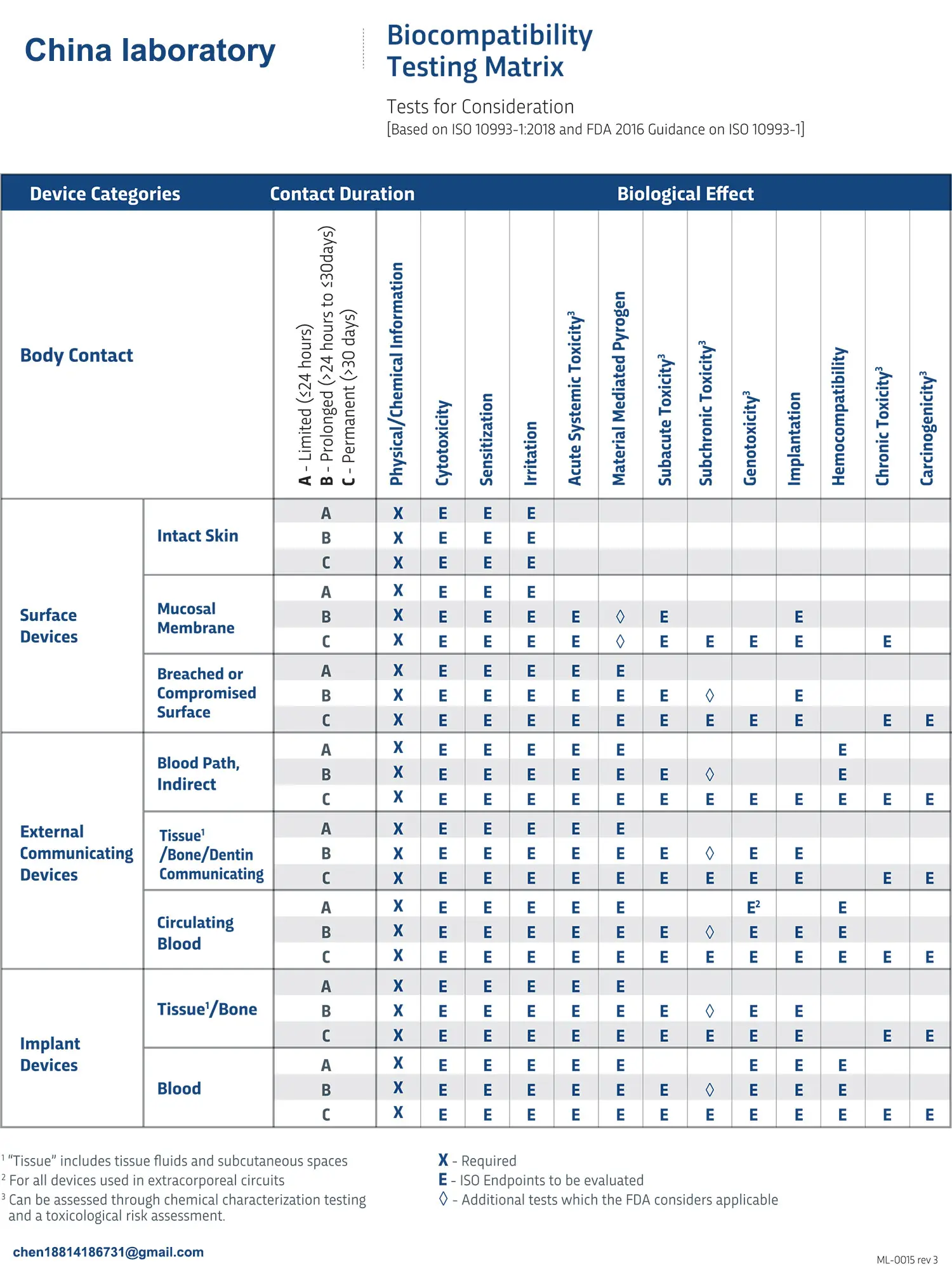 ISO 10993:5 Cytotoxicity Testing Methods
FDA ISO 10993-1 Biocompatibility Evaluation Guidel
ISO 10993:5 Cytotoxicity Testing Methods
FDA ISO 10993-1 Biocompatibility Evaluation Guidel
 In Vitro Cytotoxicity Testing for Medical Devices
In Vitro Cytotoxicity Testing for Medical Devices
Leave us a message
24-hour online customer service at any time to respond, so that you worry!