





EMC Standards for Medical Devices
Europe and the US have set industry benchmarks with their stringent certification and testing standards for medical device products sold globally, setting an industry standard by adhering to them rigorously and comprehensively. Not only are these regulations about product quality and safety; they're also critical in terms of market access for medical device makers who hope to gain entry to these markets; for them understanding and complying with US/European certification regulations is the key to succeeding here.
U.S. FDA Certification Standards
Food and Drug Administration (FDA) policies impose stringent oversight over medical devices, with certification and testing standards designed to guarantee their safety, effectiveness and regulatory compliance.
1. Device Classification and Standards Compliance (DCSC).
The Food and Drug Administration classifies medical devices by risk level into three classes, Class I for low risk devices; Class II (moderate risk devices); and Class III devices with higher risks.
Class III (High Risk)
Each classification of products falls under different regulations, such as 21 CFR Parts 800-898. Manufacturers must correctly classify their product(s), to ensure application documents comply with relevant rules (for instance: 21 CFR Part 800-898)
Class I medical cotton swabs fall under basic regulations while Class III pacemakers require compliance with more stringent specifications.
2. Design Control and Performance Verification
Manufacturers must demonstrate an extensive design control process from inputs to outputs and validate device performance through design validation. For instance: A blood glucose meter needs to be verified against standards for measurement accuracy and precision to guarantee reliable patient test results.
3 Material and Biocompatibility Evaluation
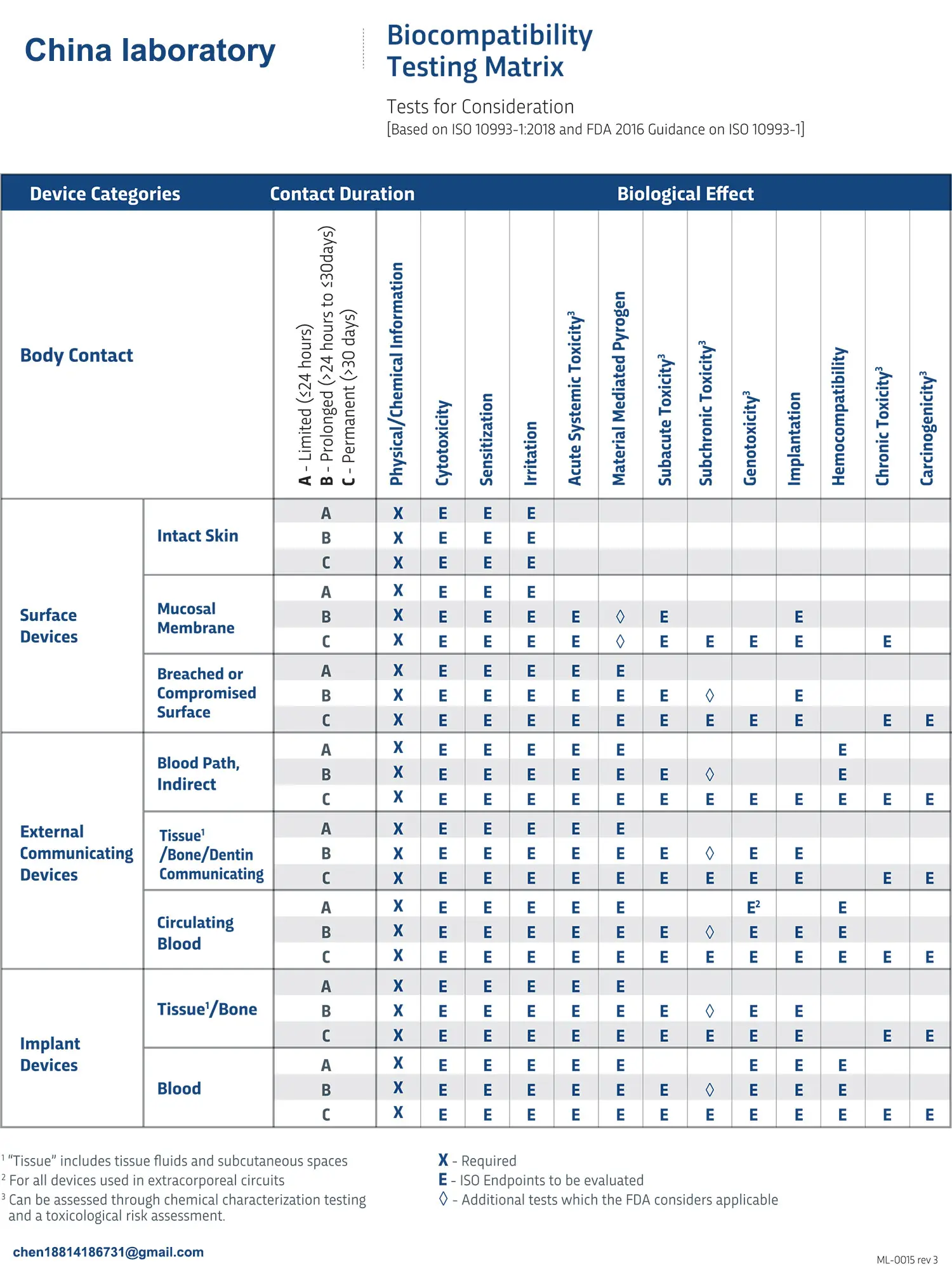
Manufacturers must submit material composition analyses and biocompatibility evaluation reports. Implantable devices also must pass tests such as cytotoxicity and sensitization in order to be safe for human bodies.
4. Risk Management
Risk Management To effectively implement risk management across its entirety, including analysis, evaluation and control strategies as well as prevention measures against errors related to disinfection errors or misuse.
5. Clinical Data and Trials
High-risk or innovative devices often require clinical trial data to ascertain safety and efficacy; such as new cardiovascular stents that must undergo large-scale clinical studies before launch.
6. Quality Management System
Establishing a quality management system compliant with ISO 13485 standards is highly recommended and should include quality planning, process control, audit records and corrective actions as key areas.
7. Declaration and Test Reports
Manufacturers must submit a declaration of conformity along with various test reports, such as IEC 60601 for electrical safety; IEC 60601-1-2 for electromagnetic compatibility (EMC); etc.
8. FDA Review and Approval Process
FDA review examines documentation to ensure it meets completeness, compliance and consistency; any additional details required will be requested from manufacturers; products can only be released into commerce after passing scrutiny successfully.
Market Surveillance and Reporting Services
The FDA mandates manufacturers submit safety data, adverse event reports and market monitoring information regularly in order to quickly detect and address issues that arise.
EU CE certification and Test Standards (Not Available for Indicator Type Products).
CE certification serves as the gateway for medical devices entering European markets, with stringent testing and certification standards covering every stage of product lifecycle development.
1. Medical Device Regulation or Directive (MDR or MDD).
On May 26th 2021, MDR officially replaced MDD; its definition includes device classification, clinical evaluation and technical documentation requirements as well as core requirements; whilst high risk devices will receive further scrutiny than lower-risk ones.
2. ISO 13485 Quality Management System
Manufacturers must establish an ISO 13485 compliant quality system covering their entire manufacturing process from raw material procurement through final delivery of goods.
3. Technical Document Requirements (TDRs)
Includes product specifications and design documents;esthetique performance and safety test reports;
biocompatibility assessments (if applicable);
clinical evaluation materials (if relevant).
4. Biocompatibility Evaluation
Comply with ISO 10993 series standards when conducting testing of device contact with human tissues/fluids for safety evaluation. Tests could include: Cytotoxicity, sensitization, subacute toxicity and chronic toxicity evaluation etc.
5.Electromagnetic Compatibility (EMC)
According to IEC 60601-1-2, devices must operate reliably in electromagnetic environments without interfering with or interferring with other equipment.
6. Clinical Evalauation of Crush Injury.
Some devices require clinical data for safety and efficacy evaluation in real world clinical use to verify they deliver expected benefits.
7 Standards and Guidelines
Certification references multiple international standards, including EN ISO 14971 (Risk Management); IEC 60601 (Electrical Safety); as well as various ISO series.
On-site Audit and Document Review Services (OTDR).
Notified Bodies will evaluate technical documentation and may conduct on-site audits to make sure actual practices align with those documented.
Market Monitoring and Reporting Solutions (MM&R)
Once they receive the CE mark, manufacturers must continue with:
Market feedback analysis and safety alert management. Ongoing performance and safety monitoring.
Conclusion Although both U.S. and EU medical device certification standards emphasize safety and efficacy, their regulatory details and review processes differ considerably;
The FDA emphasizes classification management and itemized review while in Europe these processes tend to follow an incremental review cycle model;
- CE employs an efficient full-lifecycle supervision model.
Manufacturers looking to break into the United States or European markets must carefully research certification and testing standards of both regions before developing application materials with precision for smooth product launches and reliable medical device solutions for patients around the globe.
Email:hello@jjrlab.com
Write your message here and send it to us
 Packaging Validation ISO 11607 Test Report
Packaging Validation ISO 11607 Test Report
 What is the ISO 11607-1 Packaging Validation Test?
What is the ISO 11607-1 Packaging Validation Test?
 How to get an ISO 11737-1 Test Report?
How to get an ISO 11737-1 Test Report?
 Orthopedic Implant Cleanliness Testing
Orthopedic Implant Cleanliness Testing
 What is ISO 10993-23:2021 Irritation Testing?
What is ISO 10993-23:2021 Irritation Testing?
 ISO 10993-23 Irritation Testing Laboratory
ISO 10993-23 Irritation Testing Laboratory
 EMI Emissions Testing
EMI Emissions Testing
 EMC Standards for Medical Devices
EMC Standards for Medical Devices
Leave us a message
24-hour online customer service at any time to respond, so that you worry!