





CE Marking Process for Medical Devices
What four aspects does CE certification include?
CE certification is a complete safety assurance system, not just taking a sample to a laboratory for inspection and approval.
Because the CE mark is a safety mark, a product that passes the CE certification must ensure that all aspects of the product, including design, production, packaging, instruction manual writing, transportation, sales, the entire effective service life of the product, and the recycling of the product after use, meet the basic requirements stipulated in the relevant laws on health, safety, and environmental protection in Europe. Therefore, if a manufacturer wants to pass the CE certification for its products, it usually needs to meet the following four requirements:
1. Before the product is put on the European market, affix the ce label to the product.
2. After the product is put on the European market, the technical files must be kept within the EU for inspection by supervisory agencies at any time.
3. For products that are found by market supervision agencies to be non-compliant with CE requirements, or products that have accidents during use but have been affixed with CE labels, remedial measures must be taken (such as temporarily removing them from the shelves or permanently removing them from the market).
4. After a product model with a CE label is put on the European market, if there are any changes or modifications to the relevant EU laws, the subsequent production of the same model products must also be changed or revised accordingly to comply with the new EU legal requirements.
ce certification process
1. Confirm the export country
2. Confirm product categories and relevant EU product directives
3. Appointment of “Authorized Representative”
4. Confirm the module required for authentication
5. Should we adopt the “self-declaration” model or “must go through a third-party certification body”?
6. Establish technical files and maintain and update them
1. Confirm the export country
If the product is exported to any of the 30 member states of the European Economic Area (EEA), including the European Union (EU) and the European Free Trade Agreement (EFTA), CE certification may be required.
2. Confirm product categories and relevant EU product directives
If the product belongs to any of the 22 categories listed here, generally speaking, CE certification is required. If a product belongs to more than one category at the same time, it must meet the requirements listed in the product directives corresponding to all categories.
Note: Certain product directives sometimes list products that are excluded from the directive.
3. Appointment of “eu authorized representative”
In order to ensure that the four requirements of the ce marking certification implementation process are met, EU law requires that manufacturers outside the 30 EEA member states must designate an EU Authorized Representative (EU Authorized Agent) in the EU to ensure the consistency of product "safety" during the circulation process and use after the product is put on the European market; technical files must be stored in the EU for inspection by supervisory agencies at any time; remedial measures must be taken for products that are found by market supervisory agencies to be non-compliant with CE requirements, or products that have accidents during use but have been affixed with CE labels. (For example, temporarily remove them from the shelves or permanently remove them from the market); if the product model with a CE label is changed or modified after it is put on the European market, the subsequent production of the same model must also be changed or revised accordingly to meet the new EU legal requirements.
4. Confirm the module required for authentication
For almost all EU product directives, the directives usually provide manufacturers with several CE certification (Conformity Assessment Procedures) modules. Manufacturers can choose the most suitable module according to their own situation. Generally speaking, CE certification modules can be divided into the following 9 basic modules:
1.Module A: internal production control
Mode A: Internal production control (self-declaration)
2.Module Aa: intervention of a Notified Body
Mode Aa: Internal production control plus 3rd party testing
3.Module B: EC type-examination
Mode B: EC type test
4.Module C: conformity to type
Mode C: Conform to the type
5.Module D: production quality assurance
Mode D: Production Quality Assurance
6.Module E: product quality assurance
Mode E: Product Quality Assurance
7.Module F: product verification
Mode F: Product Verification
8.Module G: unit verification
Mode G: Unit Validation
9.Module H: full quality assurance
Mode H: Comprehensive Quality Assurance
Based on the different combinations of the above basic modes, several other different modes may be derived. Generally speaking, not any mode is applicable to all products. In other words, manufacturers cannot arbitrarily choose any of the above modes to obtain CE certification for their products.
5. Should we adopt the “self-declaration” model or “must go through a third-party certification body”?
Risk Level: Minimal Risk
The EU product directive allows manufacturers of certain categories of products with a low risk level (Minimal Risk) to choose to obtain CE certification in the form of Mode A: "Internal Production Control (Self-Declaration)".
Products with higher risk levels must be certified by a third-party certification body, NB (Notified Body).
For products with a higher risk level, their manufacturers must choose other modes besides Mode A, or Mode A plus other modes to achieve CE certification. In other words, a third-party certification body NB (Notified Body) must be involved.
In the certification process of other modes other than Mode A, at least one EU-recognized certification body NB is usually required to participate in part or all of the certification process. According to different modes, NB may intervene in the certification process in the following ways: sample testing, sampling testing, factory inspection, annual inspection, different quality system audits, etc., and issue corresponding test reports, certificates, etc.
At present, more than 1,200 certification bodies have been recognized by the EU, and most of these certification bodies are located in EU member states. Usually, an NB is only authorized by the EU to certify one or several modes for a certain type or several types of products. In other words, an EU-authorized certification body cannot certify all types of products, and even for authorized product types, it is usually not authorized for all modes. For each EU product directive, there is usually a list of authorized certification bodies for that product directive.
6. Establish technical files and maintain and update them
EU law requires that after a product with a CE label is put on the European market, its technical files must be stored in the EU for inspection by supervisory agencies at any time. If there are any changes in the content of the technical files, the technical files should also be updated in a timely manner.
"Technical documentation" is a very important issue in the EU Medical Device Directive. Its purpose is to require companies to prepare sufficient technical information and certification for random inspections by competent authorities or for use in litigation disputes. The requirements for "technical files" in various EU directives are different. Here, I would like to take the requirements for "medical devices" most commonly used by Chinese export companies as an example to illustrate.
Medical Device Directive 93/42/EEC requires that the “Technical File” may include the following items:
A. The company's quality manual and procedure documents
B. Company profile and European agent name and contact information
C. CE Declaration of Conformity (or self-assurance statement. If the product is used in conjunction with other equipment, there should be evidence that the overall product meets the basic requirements)
1. Brief description of product name, classification and referenced standard clauses
2. Product overview (including type and intended use)
a) Product history
b) Technical performance parameters
c) List of accessories, matching parts and other equipment used with the product
d) Product illustrations and samples
e) Raw materials and suppliers used in products
3. Use the harmonized standard/or other standards of the product
4. Risk analysis and assessment conclusions and preventive measures (EN1441 Product Service Hazard Analysis Report)
5. Production quality control
a) Product information and control documents (including product production process flow chart)
b) Description of the product sterilization method and validation
c) Sterilization Validation
d) Product quality control measures
e) Description of product stability and shelf life
6. Packaging and labeling
a) Packaging material description
b) Tags
c) User Manual
7. Technical evaluation
a) Product inspection report and related documents
b) Technical overview and authoritative opinions
8. Potential risk assessment
a) Product potential risk test report and related literature
b) Summary of potential risks and authoritative opinions
9. Clinical evaluation
a) Product clinical test reports and related literature
b) Overview of clinical use and authoritative opinions
Appendix 1. Product factory inspection report
Appendix 2. Product Type Test Report
Appendix 3. Basic Requirements Checklist
Note:
1. Clinical research (including: physical properties, biochemistry, pharmacology, pharmacokinetic and toxicity studies, efficacy testing, sterilization certification, drug compatibility, etc.)
2. biocompatibility test
(A) EN30993 Part 1 requirements: cytotoxicity, photosensitivity, irritation - intradermal reaction, acute systemic toxicity, pyrogenicity, subacute toxicity, genotoxicity, implantation hemolysis;
(B) Supporting tests: chronic toxicity, carcinogenicity, reproductive/growth toxins, biodegradation.)
3. Clinical data (clinical research or description of clinical research is required)
4. Packaging certificate (EN868)
5. Labels, instructions for use (EN980, EN1041)
6. Conclusion (acceptance of design dossier, statement of benefits versus risks)
All the above documents must be written in one of the official languages of the European Union (English, German, French), but the instructions must be written in the language of the user's country. All documents should be kept for at least five years after the last shipment.
In vitro diagnostic medical devices IVDD product categories
According to Annex 2 of Directive 98/79/EC (IVDD), the product classification principle is determined to classify products that require certification. The basis for classification is the disease diagnosed by the product.
The calibrators, instruments, and specimen collection and preservation equipment used in conjunction with the above-mentioned diagnostic reagents all fall within the scope of the In Vitro Diagnostic Device Directive.
Medical Devices MDD Product Categories
In the CE certification process, it is not enough to judge the correct classification of a medical device based on the name of the device. The complete intended purpose must be known!
We often hear questions like this: What category of medical device does a certain product belong to in the CE classification? The questioner may not know that it is often inappropriate to judge the classification of a medical device in the CE certification process based solely on its name!
1. Differences between the EU and the US The classification of medical devices in the EU and the US is very different.
The US FDA has classified medical devices according to their common characteristics and established a public database for query;
The EU has established a set of classification rules, allowing manufacturers to classify products according to their intended use (Intended Purpose).
2. The same product can be a medical device or not a medical device
In the United States, whether a product is a medical device is determined solely by the FDA;
In the EU, whether a product is a medical device is determined by the manufacturer (declared intended use of the product). For example, an electric heating pad can be a medical device or not.
3. The same product can be different types of medical devices
For example: Depending on the intended use stated by the manufacturer, an electric heating mattress can be either a Class I medical device or a Class IIa or IIb medical device.
4. The same product belongs to different categories when it is part of a system and when it is an accessory
For example, a container for ascitic fluid retained outside the body during surgery with a non-active ascitic fluid extraction device would be class IIa when part of the system but would be class I when an accessory.
5. Similar products can be different categories of medical devices
For example: Picture Archiving and Communication Systems (PACS), an image storage and communication system commonly used in X-ray films, has different intended uses (functions) declared by different manufacturers. PACS can be a Class I medical device, or a Class IIa or IIb medical device.
6. Some similar products are medical devices (MD) and some are in vitro diagnostic devices (IVD).
For example, if blood collection tubes are invasive or in contact with the skin, they are (ordinary) medical devices MD governed by the MDD 93/42/EEC Directive.
If it is non-invasive or does not come into contact with the skin at all, it is an in vitro diagnostic device (IVD) governed by Directive 98/79/EC.
Annex IX of the Medical Device Directive MDD 93/42/eec specifies 18 rules, which classify medical products into Class I, Class IIa, Class IIb and Class III according to their degree of danger.
Product classification rules:
1. The application of the rules is determined by the intended use of the device;
2. If the device is used in conjunction with other devices, the classification rules apply to each device separately;
3. Accessories can be classified separately from other instruments used together;
4. The software that starts or affects a device is of the same type as the device.
Classification criteria:
Duration: temporary (<60 minutes), short-term (<30 days), long-term (>30 days)
Traumatic: non-traumatic, through-aperture trauma, surgical trauma, implantation.
Applicable locations: central circulation, central nervous system, and other places.
Energy supply: passive, active.
Rules 1-4: All non-invasive devices belong to Class I unless they:
For storage of body fluids (except blood bags) Class II a
Active medical devices of class Ila or higher class IIa
Changes in body fluid composition Class IIa/IIb
Some wound dressings Category II a / II b
Rule 5: Medical devices that invade human body orifices
Temporary use (dental compression materials, examination gloves) Class I
Short-term use (catheters, contact lenses) Class II a Long-term use (normal dental floss) Class II b
Rule 6-8: Surgical Invasive Devices
Reusable surgical instruments (forceps, axes) Class I
Temporary or short-term use (suture needles, surgical gloves) Class 11a
Long-term use (prosthesis, intraocular lens) Class II b
Devices contacting the central circulatory system (CCS) or central nervous system Class III
Rule 9. Therapeutic devices that give or exchange energy Class II a
(Muscle stimulator, electric drill, skin phototherapy machine, hearing aid)
Category II b working in a potentially hazardous manner
(Infant incubator, high-frequency electrosurgical unit, ultrasonic lithotripter, X-ray machine)
Rule 10. Diagnostic Equipment
Provide energy (MRI, ultrasound) Class IIa
Diagnosis/monitoring of radiopharmaceutical distribution in vivo Class II a
(r camera, positron emission imager)
Diagnosis/monitoring of physiological functions (ECG, EEG) Class II a
Monitoring of physiological functions in hazardous situations Category II b
(Blood gas analyzer during surgery)
Emitting ionizing radiation (X-ray diagnostics) Class II b
Rule 11 Active devices for controlling the passage of drugs or other substances into or out of the human body Class II a
(Suction equipment, supply pump)
If working in a potentially hazardous manner Category II b
(anesthesia machine, ventilator, dialysis machine, hyperbaric oxygen chamber)
Rule 12 All other active medical devices are classified as Class I
(Observation lamps, dental chairs, wheelchairs, dental treatment lamps, active devices for recording, processing, observing and diagnosing images)
Rule 13. Devices combined with medicinal substances (condoms containing spermicide, endodontic materials containing antibiotics) Class III
Rule 14. Contraceptive devices (condoms, diaphragms, class II b) and class II b/III (intrauterine contraceptive devices, class III)
Rule 15. Cleaning or sterilizing equipment
Medical device (endoscope disinfection) Class II a
Contact lenses (disinfectants, care solutions) Class II a
Rule 16. Devices for recording X-ray images (X-ray films) Class II a
Rule 17. Devices using animal tissue (biological heart valves, gut, collagen) Class III
Rule 18, Blood Bags Category II b
CE certification steps for various types of medical devices
CE certification steps for Class I medical devices
1. Classification: Confirm that the product belongs to Class I medical device
2. Select the conformity assessment method: please refer to the flowchart below
3. Prepare technical documents
4. CE Declaration of Conformity
5. Appointment of EU Authorized Representative
6. The EU authorized representative shall register the manufacturer and product with the EU competent authority
7. Establish a post-sale alert system/ affix CE label and place the product on the market
Class I Medical Devices: ce conformity Assessment Pathway
1. Manufacturers are responsible for ensuring that their products comply with all relevant essential requirements of Directive 93/42/eec and must make a written (self) declaration to ensure this.
2. Class I medical devices without measurement functions or non-sterile (CE certification process) do not require the participation of a third-party Notified Body (NB). Whether it complies with the ISO13485:2003 standard is a voluntary choice by the manufacturer and is not mandatory.
3. Class I medical devices with measuring functions or sterilization functions must have the participation of a third-party notified body (NB) during the CE certification process.
4. Once a manufacturer believes that its product meets all relevant essential requirements of Directive 93/42/EEC, the manufacturer (within the EU) or the EU authorized representative (for manufacturers outside the EU) must first register with the EU competent authority before affixing the CE label and placing the product on the EEA market.
")
CE certification steps for Class IIa medical devices
1. Classification: Confirm that the product belongs to Class IIa medical device
2. Select the conformity assessment method: please refer to the flowchart below
3. Prepare technical documents
4. Appointment of EU Authorized Representative
5. Obtain ce certificate from a third-party notified body (NB)
6. (Complete) CE Declaration of Conformity
7. Keep the technical documents at the EU authorized representative office (for inspection by EU competent authorities at any time)
8. Establish a (after-sales) alert system/affix CE label and place the product on the EEA market
")
CE certification steps for Class IIb medical devices
1. Classification: Confirm that the product belongs to Class IIb medical device
2. Select the conformity assessment method: please refer to the flowchart below
3. Prepare technical documents
4. Appointment of EU Authorized Representative
5. Obtain CE certificate from a third-party notified body (NB)
6. (Complete) CE Declaration of Conformity
7. Keep the technical documents at the EU authorized representative office (for inspection by EU competent authorities at any time)
8. Establish a (after-sales) alert system/affix CE label and place the product on the EEA market
")
CE certification steps for Class III medical devices
1. Classification: Confirm that the product belongs to Class III medical device
2. Select the conformity assessment method: please refer to the flowchart below
3. Prepare technical documents
4. Appointment of EU Authorized Representative
5. Obtain CE certificate from a third-party notified body (NB)
6. (Complete) CE Declaration of Conformity
7. Keep the technical documents at the EU authorized representative office (for inspection by EU competent authorities at any time)
8. Establish a (after-sales) alert system/affix CE label and place the product on the EEA market
")
Email:hello@jjrlab.com
Write your message here and send it to us
 What is CPSC eFiling
What is CPSC eFiling
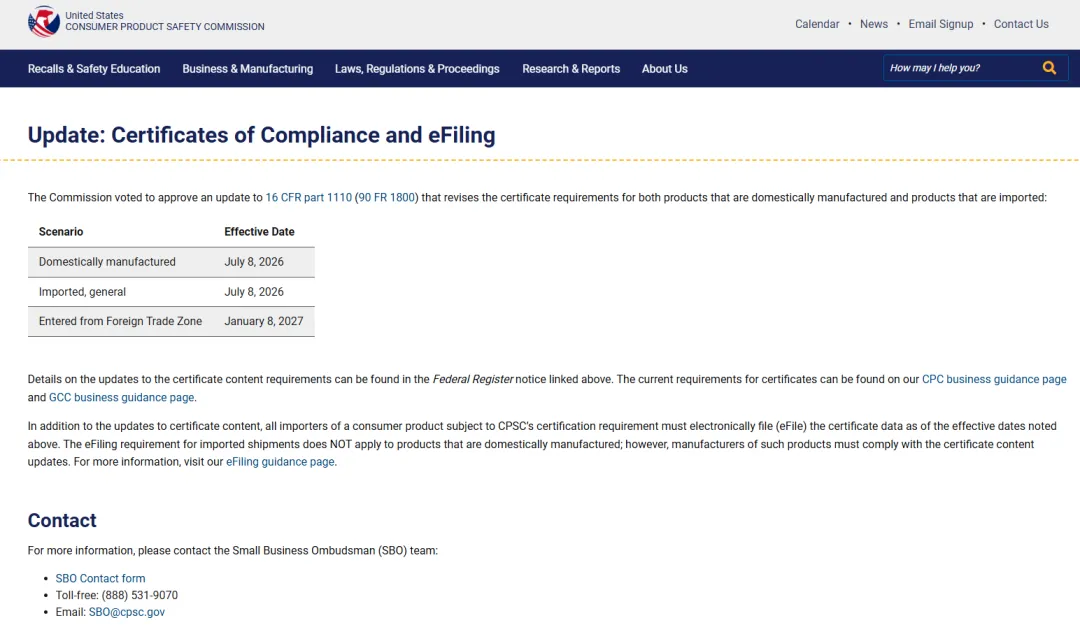 CPSC Products Must Undergo eFiling
What is the U.S. CPSC eFiling System?
CPSC Products Must Undergo eFiling
What is the U.S. CPSC eFiling System?
 IMPORTANT: Amazon US CPSC Certificates Must Be eFi
IMPORTANT: Amazon US CPSC Certificates Must Be eFi
 ROHS and Weee Compliance
ROHS and Weee Compliance
 Waste of Electrical and Electronic Equipment Weee
Waste of Electrical and Electronic Equipment Weee
 Low Voltage Directive CE Marking
Low Voltage Directive CE Marking
 What are CE EMC Testing Requirements
What are CE EMC Testing Requirements
Leave us a message
24-hour online customer service at any time to respond, so that you worry!