





CE Mark Approval for Medical Devices
What is the CE Mark for Medical Devices?
To sell medical devices in the European Union (EU), products must obtain the CE mark. This mark signifies that the medical device complies with applicable EU regulations.
")
Steps to Obtain a CE Mark
1. Identify the Target Market
If exporting to any of the 30 member countries in the European Economic Area (EEA), which includes the EU and the European Free Trade Agreement (EFTA) nations such as Sweden, Norway, Austria, and Portugal, CE certification is likely required.
2. Determine Product Classification and Applicable EU Directives
Medical devices are categorized into four classes based on risk level under the European framework: Class I, Class IIa, Class IIb, and Class III. Class III devices carry the highest risk.
- Class I Medical Devices:
Low-risk devices. Manufacturers can choose one of three CE marking routes. Class I devices are further defined as:
- Sterile devices, e.g., personal protective kits.
- Devices with a measuring function, e.g., stethoscopes.
- Non-sterile and non-measuring devices, e.g., corrective eyeglasses.
- Class IIa Medical Devices:
Medium-to-low-risk devices for short-term use (not exceeding 30 days), such as surgical gloves, hearing aids, and diagnostic ultrasound machines.
- Class IIb Medical Devices:
Medium-to-high-risk devices, including long-term corrective contact lenses, surgical lasers, and defibrillators. These are typically used for more than 30 days.
- Class III Medical Devices:
High-risk devices requiring continuous monitoring throughout their lifecycle. Examples include cardiovascular catheters, aneurysm clips, hip implants, and artificial heart valves.
3. Appoint an EU Authorized Representative
Manufacturers outside the EEA must designate an Authorized Representative within the EU to ensure compliance with CE marking requirements.
---
4. Select the Appropriate Conformity Assessment Module
Most EU directives offer manufacturers various CE certification models, or "modules," allowing them to choose the one most suitable for their situation. These include:
1. Module A: Internal production control (self-declaration).
2. Module Aa: Internal production control with third-party intervention.
3. Module B: EC type-examination.
4. Module C: Conformity to type.
5. Module D: Production quality assurance.
6. Module E: Product quality assurance.
7. Module F: Product verification.
8. Module G: Unit verification.
9. Module H: Full quality assurance.
Custom combinations of these modules may also be applicable based on product specifics. Not all modules are suitable for every product, and manufacturers cannot arbitrarily select a module.
5. Choose Between Self-Declaration or Third-Party Certification
- Low-Risk Products:
For minimal-risk products, manufacturers may opt for Module A (self-declaration).
- Higher-Risk Products:
Products with higher risk levels require third-party intervention by a Notified Body (NB). These assessments can include sample testing, factory audits, annual inspections, or quality system evaluations, depending on the selected module.
6. Prepare and Maintain Technical Documentation
EU laws mandate that products bearing the CE mark must have technical documentation stored within the EU for regulatory inspection. This documentation must be updated as changes occur.
For medical devices, the technical file typically includes:
- Quality Manual and Procedures
- Company Profile and EU Authorized Representative Information
- CE Declaration of Conformity, including:
- Product name, classification, and applicable standards.
- Product overview, intended use, and historical background.
- Technical specifications, components, and raw materials.
- Risk analysis and mitigation measures.
- Manufacturing process flowchart and quality control measures.
- Sterilization methods and validation.
- Stability and shelf-life data.
- Packaging and Labeling
- Description of packaging materials.
- Labels and instructions for use.
By following these steps and fulfilling the required documentation and assessment criteria, manufacturers can achieve CE mark approval for their medical devices.
Email:hello@jjrlab.com
Write your message here and send it to us
 Toothbrush FDA Certification Testing
Toothbrush FDA Certification Testing
 Snoring Device FDA 510k Standard Testing
Snoring Device FDA 510k Standard Testing
 Single Use Intravenous Catheter Certification Test
Single Use Intravenous Catheter Certification Test
 Silicone Material Product Compliance Certification
Silicone Material Product Compliance Certification
 What to Do If Cytotoxicity Test Results Are Positi
What to Do If Cytotoxicity Test Results Are Positi
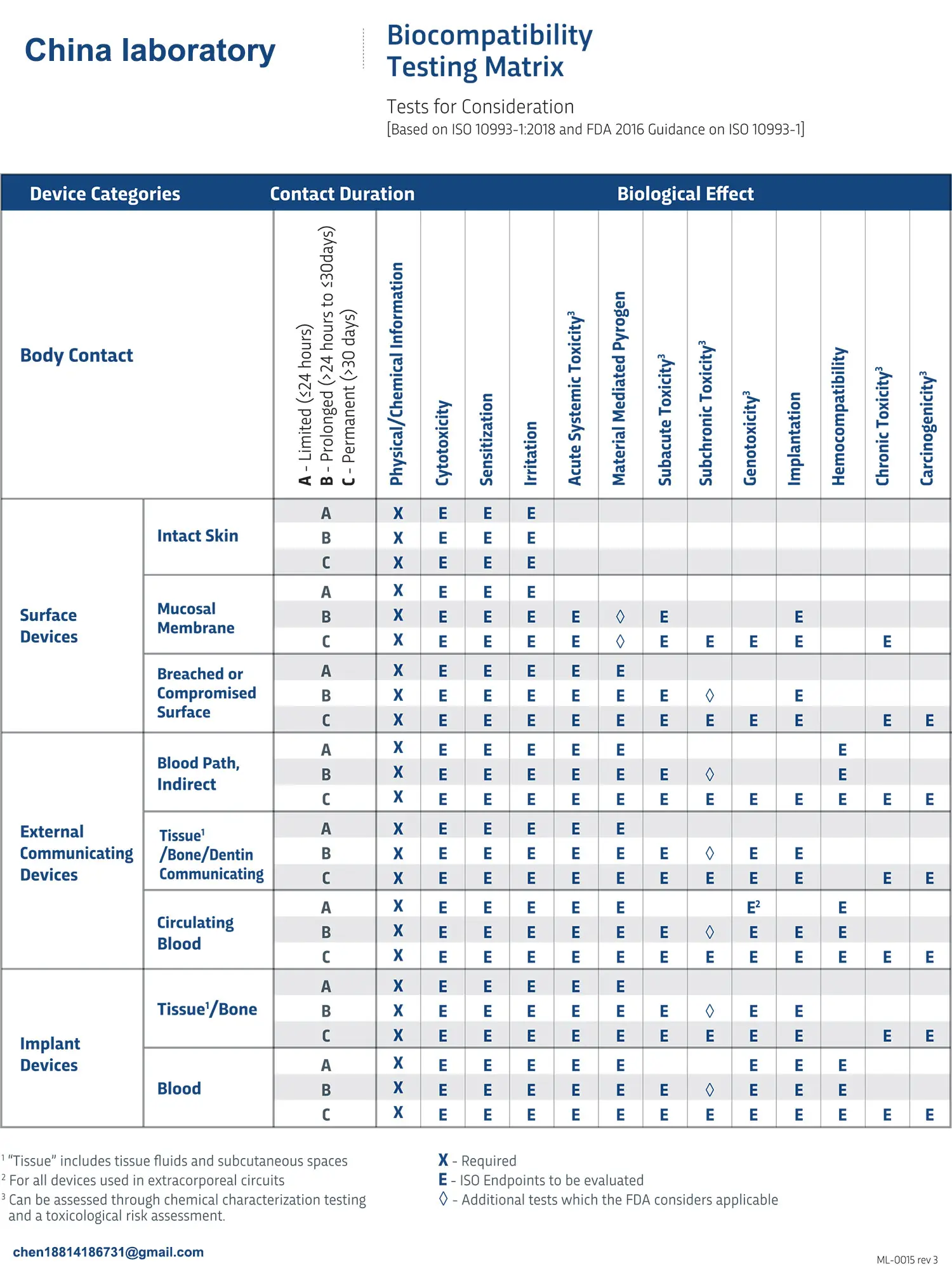 ISO 10993:5 Cytotoxicity Testing Methods
ISO 10993:5 Cytotoxicity Testing Methods
 FDA ISO 10993-1 Biocompatibility Evaluation Guidel
FDA ISO 10993-1 Biocompatibility Evaluation Guidel
 In Vitro Cytotoxicity Testing for Medical Devices
In Vitro Cytotoxicity Testing for Medical Devices
Leave us a message
24-hour online customer service at any time to respond, so that you worry!