





CE Certification for Beauty Medical Devices
What is CE certification for Medical Devices?
CE certification is a safety certification of the European Union. All medical devices entering the EU market must undergo CE certification for medical devices. The CE directives that medical devices need to meet include the Active Implantable Medical Devices Directive (AIMDD, 90/385/EEC), the Medical Devices Directive (MDD, 93/42/EEC), and the In Vitro Diagnostic Medical Devices Directive (IVDD, 98/79/EC).
Safety certification mark edited as follows for obtaining CE certification:
For a product to successfully obtain CE certification, three aspects need to be addressed:
First, collect relevant EU technical regulations and EU (EN) standards related to certified products and incorporate them into the company's product standards through digestion, absorption, and integration.
Second, the company must strictly organize production in accordance with the above product standards, meaning that the requirements of the technical regulations and EN standards should be implemented throughout the entire process of product design, development, and manufacturing.
Third, the company must establish and maintain a quality system in accordance with ISO9000+ISO13485 standards and obtain ISO9000+ISO13485 certification.
Medical Device CE Marking Process:
Currently, some domestic medical device manufacturers (especially some township enterprises) lack understanding of medical device directives and do not know how to apply for CE marking. To address this, we will briefly introduce the steps required to obtain CE marking as follows:
Step One: Analyze the device and its characteristics to determine whether it falls within the scope of the directive. The definition of medical devices is clearly stipulated in the directive. Some products, such as massage devices and masks, may seem like medical devices but are not actually within the scope of the medical device directive.
Step Two: Confirm the applicable essential requirements. The directive stipulates that any medical device must meet the intended purpose specified in Appendix I. Therefore, for manufacturers, the first and most important thing to do is to confirm all the basic requirements applicable to their products.
Step Three: Confirm any relevant European harmonized standards. Harmonized standards are standards published in the official journal of the European Union by the European Committee for Standardization (CEN) and the European Committee for Electrotechnical Standardization (CENELEC). For a certain medical device, there may be multiple harmonized standards applicable to it. Therefore, great care should be taken in confirming which harmonized standards apply to a particular product.
Step Four: Ensure that the product meets the requirements of the essential requirements or harmonized standards and document the evidence. Manufacturers should be able to provide sufficient evidence (such as testing conducted by notified bodies or other testing institutions based on harmonized standards) to prove that the product complies with the essential requirements.
Step Five: Product classification. According to the classification rules in Appendix IX of the directive, medical devices are divided into four classes: Class I, Class IIa, Class IIb, and Class III. Different types of products have different paths (conformity assessment procedures) to obtain CE marking, so it is crucial for manufacturers to accurately determine the classification of their products.
Step Six: Determine the corresponding conformity assessment procedure. For manufacturers of Class IIa, Class IIb, and Class III medical devices, there is the question of how to choose the approach for conformity assessment. The main difference lies in whether to choose the method of type testing or the method of quality system. Each approach has its characteristics, and manufacturers should choose the most suitable approach based on their actual situation.
Step Seven: Select a notified body. For Class IIa, Class IIb, and Class III medical devices, as well as sterile or measuring function Class I medical devices, a notified body should be selected, and a conformity assessment procedure should be carried out. The list of notified bodies published in the official journal of the European Union specifies strict regulations on the types of medical device certifications and conformity assessment procedures that each notified body can undertake. Therefore, manufacturers must be very cautious when choosing a notified body to avoid unnecessary losses.
Step Eight: Draft a Declaration of Conformity and affix the CE marking. The Declaration of Conformity is an important document. Each type of device must include a Declaration of Conformity as described in the appendices of the medical device directive.
Technical Documents for Medical Device CE Certification:
1. Product instructions for use.
2. Product technical specifications (or company standards), establishing technical documentation.
3. Product electrical schematics, circuit diagrams, and block diagrams.
4. List of key components or raw materials (please use products with European certification marks).
5. Copies of certification for the entire machine or key components.
6. Other required documents."
Email:hello@jjrlab.com
Write your message here and send it to us
 ASTM D4169 Drop Test
ASTM D4169 Drop Test
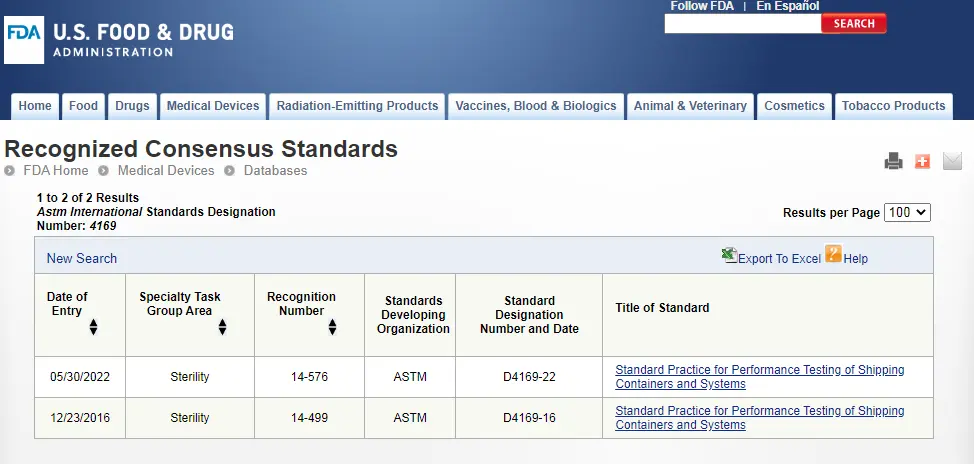 ASTM D4169 Packaging Simulation Transportation Tes
ASTM D4169 Packaging Simulation Transportation Tes
 What is ASTM D4169 Testing?
What is ASTM D4169 Testing?
 ASTM D4169-23 Test Standard Revision
ASTM D4169-23 Test Standard Revision
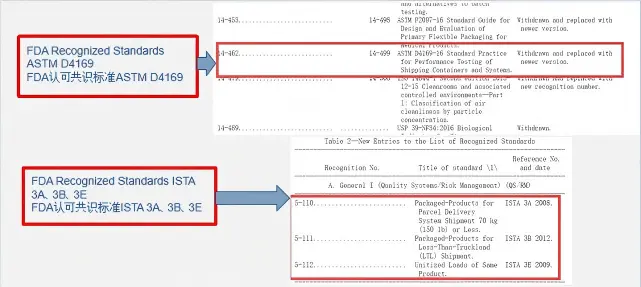 Transport Simulation Testing for Medical Device Pa
Transport Simulation Testing for Medical Device Pa
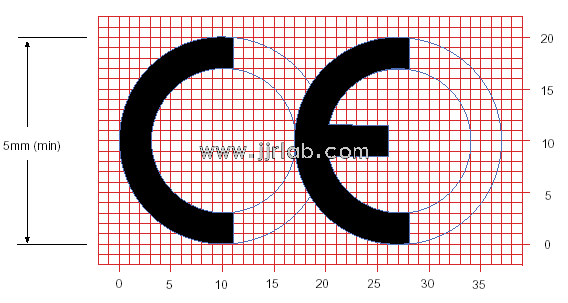 EU CE Certification Guidelines for Lighting Fixtur
EU CE Certification Guidelines for Lighting Fixtur
 Lithium Battery Export: CB Certification & IEC
Lithium Battery Export: CB Certification & IEC
 How to Apply for One FCC Certificate for Multiple
How to Apply for One FCC Certificate for Multiple
Leave us a message
24-hour online customer service at any time to respond, so that you worry!